Circa 2021 force field this can also shield the earth or cities.
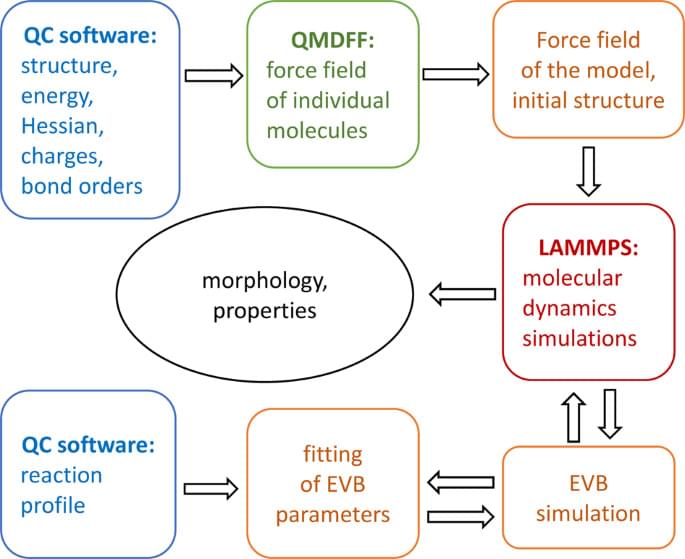
The computational design of functional materials relies heavily on large-scale atomistic simulations. Such simulations are often problematic for conventional classical force fields, which require tedious and time-consuming parameterization of interaction parameters. The problem can be solved using a quantum mechanically derived force field (QMDFF)—a system-specific force field derived directly from the first-principles calculations. We present a computational approach for atomistic simulations of complex molecular systems, which include the treatment of chemical reactions with the empirical valence bond approach. The accuracy of the QMDFF is verified by comparison with the experimental properties of liquid solvents.